
Planemo workflow
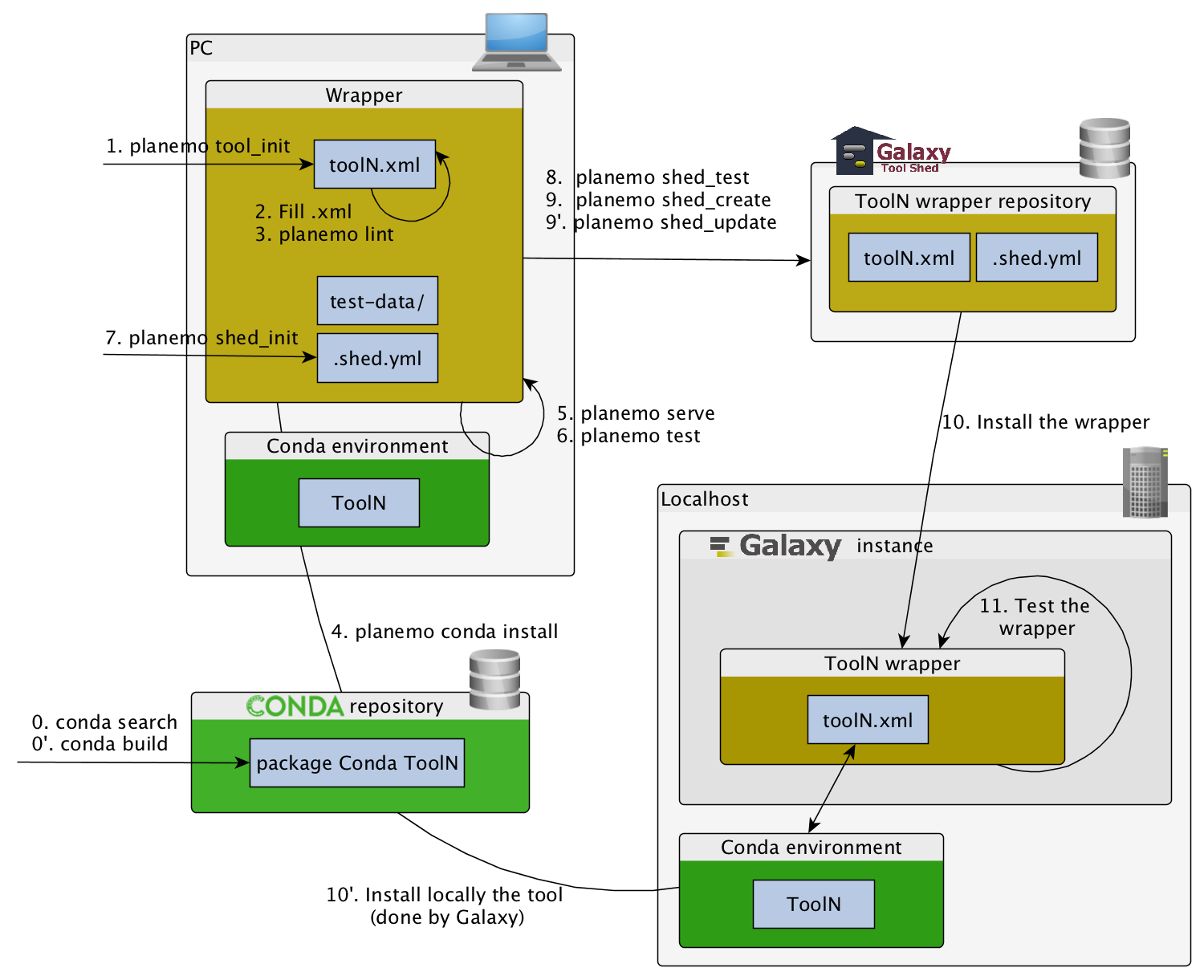
planemo tool_init命令
创建xml文件的基本骨架
$ mkdir new_tool #创建new_tool文件夹
$ cd new_tool
$ planemo tool_init --id 'some_short_id' --name 'My super tool' #创建xml文件名为some_short_id, name为My super tool
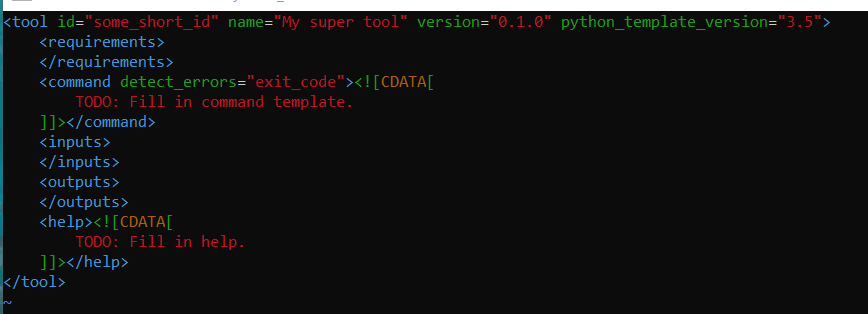
该文件内容如下:
对于planemo tool_init更多的选项指令:
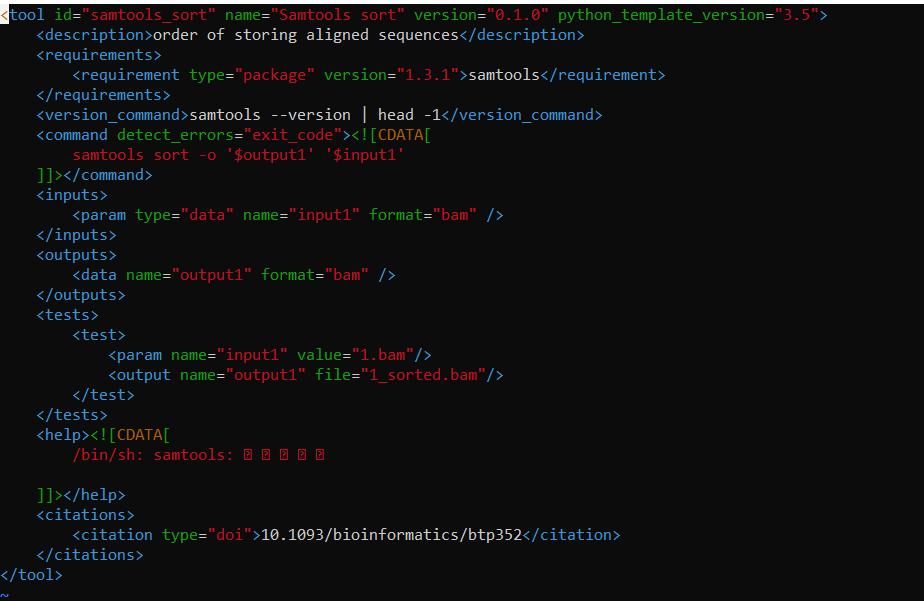
如果使用下面的代码建立xml文件:
planemo tool_init --id 'samtools_sort' --name 'Samtools sort' \
--description 'order of storing aligned sequences' \
--requirement 'samtools@1.3.1' \
--example_command "samtools sort -o '1_sorted.bam' '1.bam'" \
--example_input 1.bam \
--example_output 1_sorted.bam \
--test_case \
--version_command 'samtools --version | head -1' \
--help_from_command 'samtools sort' \
--doi '10.1093/bioinformatics/btp352'
该文件的内容就是:
使用代码planemo tool_init --help可以查看详细指令情况:
Usage: planemo tool_init [OPTIONS]
Generate tool outline from given arguments.
Options:
-i, --id TEXT Short identifier for new tool (no whitespace)
-f, --force Overwrite existing tool if present.
-t, --tool FILE Output path for new tool (default is <id>.xml)
-n, --name TEXT Name for new tool (user facing)
--version TEXT Tool XML version.
-d, --description TEXT Short description for new tool (user facing)
-c, --command TEXT Command potentially including cheetah variables
()(e.g. 'seqtk seq -a $input > $output')
--example_command TEXT Example to command with paths to build Cheetah
template from (e.g. 'seqtk seq -a 2.fastq >
2.fasta'). Option cannot be used with --command,
should be used --example_input and
--example_output.
--example_input TEXT For use with --example_command, replace input file
(e.g. 2.fastq with a data input parameter).
--example_output TEXT For use with --example_command, replace input file
(e.g. 2.fastq with a tool output).
--named_output TEXT Create a named output for use with command block
for example specify --named_output=output1.bam and
then use '-o $output1' in your command block.
--input TEXT An input description (e.g. input.fasta)
--output TEXT An output location (e.g. output.bam), the Galaxy
datatype is inferred from the extension.
--help_text TEXT Help text (reStructuredText)
--help_from_command TEXT Auto populate help from supplied command.
--doi TEXT Supply a DOI (http://www.doi.org/) easing citation
of the tool for Galxy users (e.g. 10.1101/014043).
--cite_url TEXT Supply a URL for citation.
--test_case For use with --example_commmand, generate a tool
test case from the supplied example.
--macros Generate a macros.xml for reuse across many tools.
--version_command TEXT Command to print version (e.g. 'seqtk --version')
--requirement TEXT Add a tool requirement package (e.g. 'seqtk' or
'seqtk@1.68').
--container TEXT Add a Docker image identifier for this tool.
--cwl Build a CWL tool instead of a Galaxy tool.
--help Show this message and exit.
planemo lint 命令
planemo lint 用来检查包装器的语法(Checks the syntax of a wrapper)。这里直接在终端输入planemo lint 来检查刚才我们用比较长的命令所建的xml文件。
Applying linter tests... CHECK
.. CHECK: 1 test(s) found.
Applying linter output... CHECK
.. INFO: 1 outputs found.
Applying linter inputs... CHECK
.. INFO: Found 1 input parameters.
Applying linter help... CHECK
.. CHECK: Tool contains help section.
.. CHECK: Help contains valid reStructuredText.
Applying linter general... CHECK
.. CHECK: Tool defines a version [0.1.0].
.. CHECK: Tool defines a name [Samtools sort].
.. CHECK: Tool defines an id [samtools_sort].
.. CHECK: Tool targets 16.01 Galaxy profile.
Applying linter command... CHECK
.. INFO: Tool contains a command.
Applying linter citations... CHECK
.. CHECK: Found 1 likely valid citations.
Applying linter tool_xsd... CHECK
.. INFO: File validates against XML schema.
planemo serve 命令
让你的工具在本地galaxy中实现,直接在终端输入:
planemo serve
然后运行的时候,我感觉好慢好慢,你看:
打开浏览器http://127.0.0.1:9090/地址查看工具。
planemo test 命令
使用planemo test测试工具的功能:
planemo test




